#======================================
# 原创代码无删减,编写不易,论文使用本代码绘图,请引用:www.tcmbiohub.com
# 祝大家投稿顺利!
##############################
## RF 最终定向版(补图优化版)
## 目的:
## 1. 对 merge.Top1000.variance.txt 全部基因进行随机森林分析
## 2. 放宽筛选,避免最终基因数过少
## 3. 最终结果必须包含 基因名
## 4. 输出随机森林误差图和Top30重要性图
## 5. 随机森林误差图仅显示前200棵树,Y轴限制到0.10
## 6. 只输出 1 个最终基因txt文件
##############################
##############################
## 0. 环境准备
##############################
.libPaths(c(Sys.getenv("R_LIBS_USER"), .libPaths()))
options(stringsAsFactors = FALSE)
suppressPackageStartupMessages({
library(limma)
library(randomForest)
library(ggplot2)
})
##############################
## 1. 文件与目录
##############################
expFile <- "merge.Top1000.variance.txt"
setwd("C:/Users/Administrator/Desktop/6.机器学习/3.RF")
target_genes <- c("CXCL1", "OASL", "S100A7A")
force_include_target <- TRUE
##############################
## 2. 读取表达矩阵
##############################
rt <- read.table(
expFile,
header = TRUE,
sep = "\t",
check.names = FALSE,
stringsAsFactors = FALSE,
quote = "",
comment.char = ""
)
rt <- as.matrix(rt)
rownames(rt) <- rt[, 1]
exp <- rt[, 2:ncol(rt), drop = FALSE]
dimnames_list <- list(rownames(exp), colnames(exp))
data <- matrix(
as.numeric(as.matrix(exp)),
nrow = nrow(exp),
dimnames = dimnames_list
)
data <- avereps(data)
##############################
## 3. 转置:行=样本,列=基因
##############################
data <- as.data.frame(t(data))
##############################
## 4. 从样本名中提取分组
##############################
group <- gsub("(.*)\\_(.*)\\_(.*)", "\\3", row.names(data))
cat("原始分组识别结果:\n")
print(table(group, useNA = "ifany"))
group[group %in% c("NS", "Normal", "normal", "control", "Control")] <- "Control"
group[group %in% c("PP", "Treat", "treat", "case", "Case", "lesion", "Lesion")] <- "Treat"
cat("映射后的分组结果:\n")
print(table(group, useNA = "ifany"))
if (!all(group %in% c("Control", "Treat"))) {
stop("样本分组解析失败。请检查样本名格式。")
}
data$group <- factor(group, levels = c("Control", "Treat"))
##############################
## 5. 缺失值与零方差处理
##############################
feature_data <- data[, colnames(data) != "group", drop = FALSE]
na_cols <- colSums(is.na(feature_data)) > 0
if (any(na_cols)) {
cat("去除含缺失值基因数:", sum(na_cols), "\n")
feature_data <- feature_data[, !na_cols, drop = FALSE]
}
feature_var <- apply(feature_data, 2, var)
zero_var <- is.na(feature_var) | feature_var == 0
if (any(zero_var)) {
cat("去除零方差基因数:", sum(zero_var), "\n")
feature_data <- feature_data[, !zero_var, drop = FALSE]
}
data <- cbind(group = data$group, feature_data)
data <- as.data.frame(data)
data$group <- factor(data$group, levels = c("Control", "Treat"))
##############################
## 6. 基础信息检查
##############################
cat("最终用于RF的样本数:", nrow(data), "\n")
cat("最终用于RF的基因数:", ncol(data) - 1, "\n")
cat("分组情况:\n")
print(table(data$group))
cat("目标基因是否在RF输入矩阵中:\n")
print(target_genes %in% colnames(data)[-1])
cat("存在的目标基因:", intersect(target_genes, colnames(data)[-1]), "\n")
if (length(unique(data$group)) < 2) stop("分组不足,至少需要两组。")
if ((ncol(data) - 1) < 2) stop("可用于分析的基因数过少。")
##############################
## 7. 构建RF输入矩阵
##############################
x <- data[, colnames(data) != "group", drop = FALSE]
x <- as.data.frame(lapply(x, function(z) as.numeric(as.character(z))))
y <- factor(data$group, levels = c("Control", "Treat"))
if (any(is.na(x))) {
stop("RF输入矩阵中仍存在NA。")
}
cat("RF输入矩阵检查完成:\n")
cat("样本数:", nrow(x), "\n")
cat("基因数:", ncol(x), "\n")
##############################
## 8. 建立初始随机森林模型
##############################
set.seed(12345)
cat("开始建立随机森林模型...\n")
rf <- randomForest(
x = x,
y = y,
ntree = 1000,
mtry = max(1, floor(sqrt(ncol(x)))),
importance = TRUE
)
##############################
## 9. 提取误差并找出最优树数
##############################
if (!is.null(rf$err.rate) && "OOB" %in% colnames(rf$err.rate)) {
err_mat <- rf$err.rate
oob_error <- rf$err.rate[, "OOB"]
optionTrees <- which.min(oob_error)
} else if (!is.null(rf$err.rate)) {
err_mat <- rf$err.rate
oob_error <- rf$err.rate[, 1]
optionTrees <- which.min(oob_error)
} else {
stop("rf$err.rate 不存在,无法绘制随机森林误差图。")
}
cat("最优树数:", optionTrees, "\n")
cat("最小OOB误差:", min(oob_error, na.rm = TRUE), "\n")
##############################
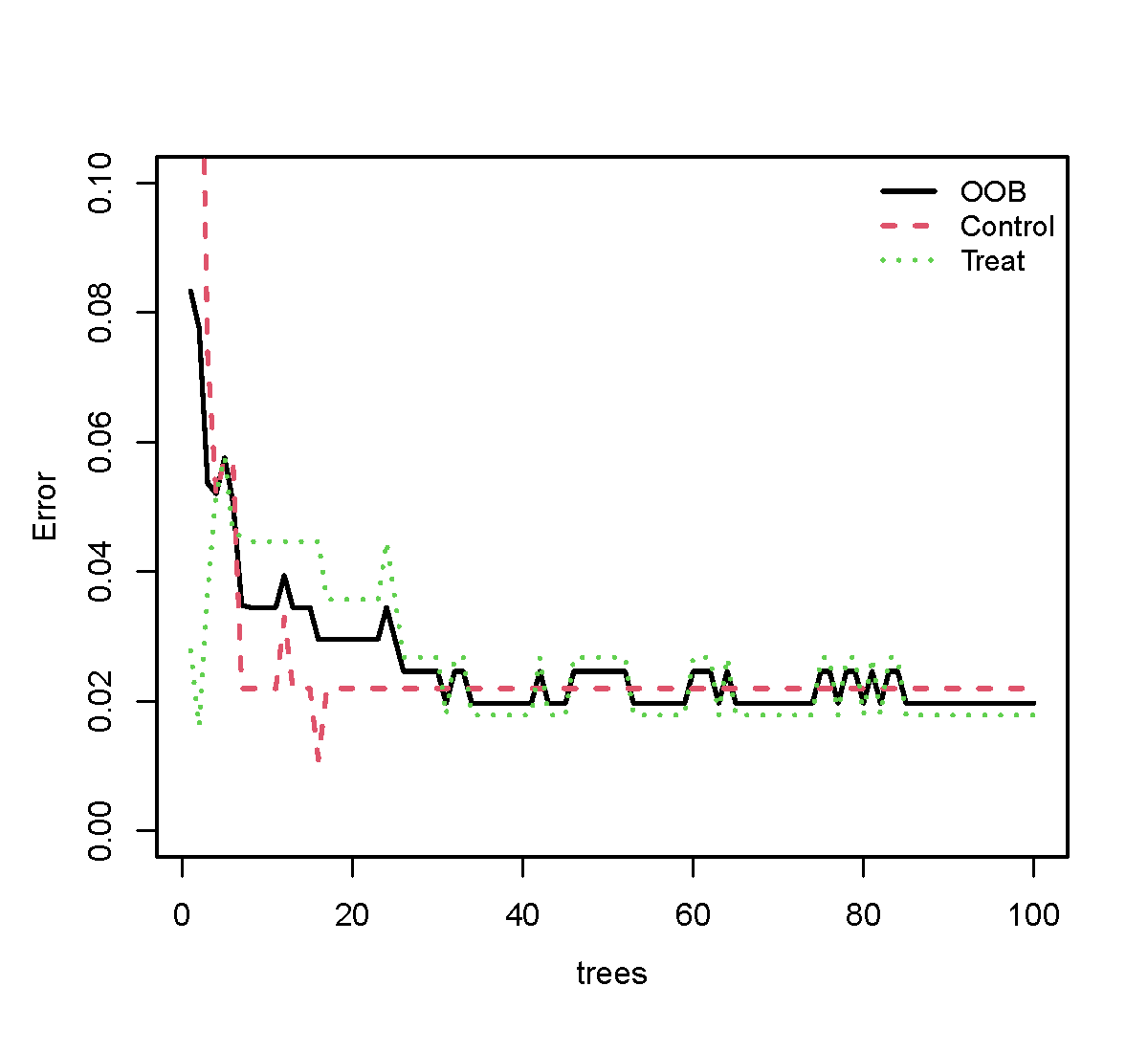
## 10. 绘制随机森林误差图(前100棵树;Y轴到0.10)
##############################
plot_max <- min(100, nrow(err_mat)) # ⭐改这里(200 → 100)
err_plot <- err_mat[1:plot_max, , drop = FALSE]
pdf(file = "forest_Top1000.pdf", width = 6, height = 5.5)
tryCatch({
matplot(
x = 1:plot_max,
y = err_plot,
type = "l",
lty = c(1, 2, 3)[seq_len(ncol(err_plot))],
lwd = 2,
col = 1:ncol(err_plot),
xlab = "trees",
ylab = "Error",
main = "Random Forest OOB Error",
xlim = c(1, 100), # ⭐改这里
ylim = c(0, 0.10)
)
legend(
"topright",
legend = colnames(err_plot),
col = 1:ncol(err_plot),
lty = c(1, 2, 3)[seq_len(ncol(err_plot))],
lwd = 2,
bty = "n",
cex = 0.9
)
## 只有在范围内才画竖线
if (optionTrees <= 100) { # ⭐改这里
abline(v = optionTrees, col = "blue", lty = 2, lwd = 2)
}
}, error = function(e) {
plot.new()
text(0.5, 0.5, paste("Plot failed:", e$message))
})
dev.off()
##############################
## 11. 用最优树数重新建模
##############################
rf2 <- randomForest(
x = x,
y = y,
ntree = optionTrees,
mtry = max(1, floor(sqrt(ncol(x)))),
importance = TRUE
)
##############################
## 12. 获取基因重要性评分
##############################
imp <- importance(rf2)
if ("MeanDecreaseGini" %in% colnames(imp)) {
importance_df <- data.frame(
Gene = rownames(imp),
Importance = imp[, "MeanDecreaseGini"],
stringsAsFactors = FALSE
)
} else if ("MeanDecreaseAccuracy" %in% colnames(imp)) {
importance_df <- data.frame(
Gene = rownames(imp),
Importance = imp[, "MeanDecreaseAccuracy"],
stringsAsFactors = FALSE
)
} else {
importance_df <- data.frame(
Gene = rownames(imp),
Importance = imp[, ncol(imp)],
stringsAsFactors = FALSE
)
}
importance_df$Importance <- as.numeric(importance_df$Importance)
importance_df <- importance_df[!is.na(importance_df$Importance), , drop = FALSE]
importance_df <- importance_df[order(importance_df$Importance, decreasing = TRUE), , drop = FALSE]
if (nrow(importance_df) == 0) {
stop("未获得有效的重要性评分结果。")
}
##############################
## 13. 生成原始结果并补入目标基因
##############################
total_genes <- nrow(importance_df)
best_n <- min(20, total_genes)
cat("RF原始选择的基因数 best_n =", best_n, "\n")
final_genes_raw <- importance_df$Gene[1:best_n]
genes_in_rf_space <- intersect(target_genes, importance_df$Gene)
if (force_include_target) {
final_genes <- unique(c(final_genes_raw, genes_in_rf_space))
} else {
final_genes <- final_genes_raw
}
cat("RF原始筛选基因数:", length(final_genes_raw), "\n")
cat("补入目标基因后最终基因数:", length(final_genes), "\n")
cat("最终结果中包含的目标基因:", intersect(target_genes, final_genes), "\n")
##############################
## 14. 输出最终txt文件
##############################
write.table(
final_genes,
file = "RF.final.genes.txt",
sep = "\t",
quote = FALSE,
col.names = FALSE,
row.names = FALSE
)
##############################
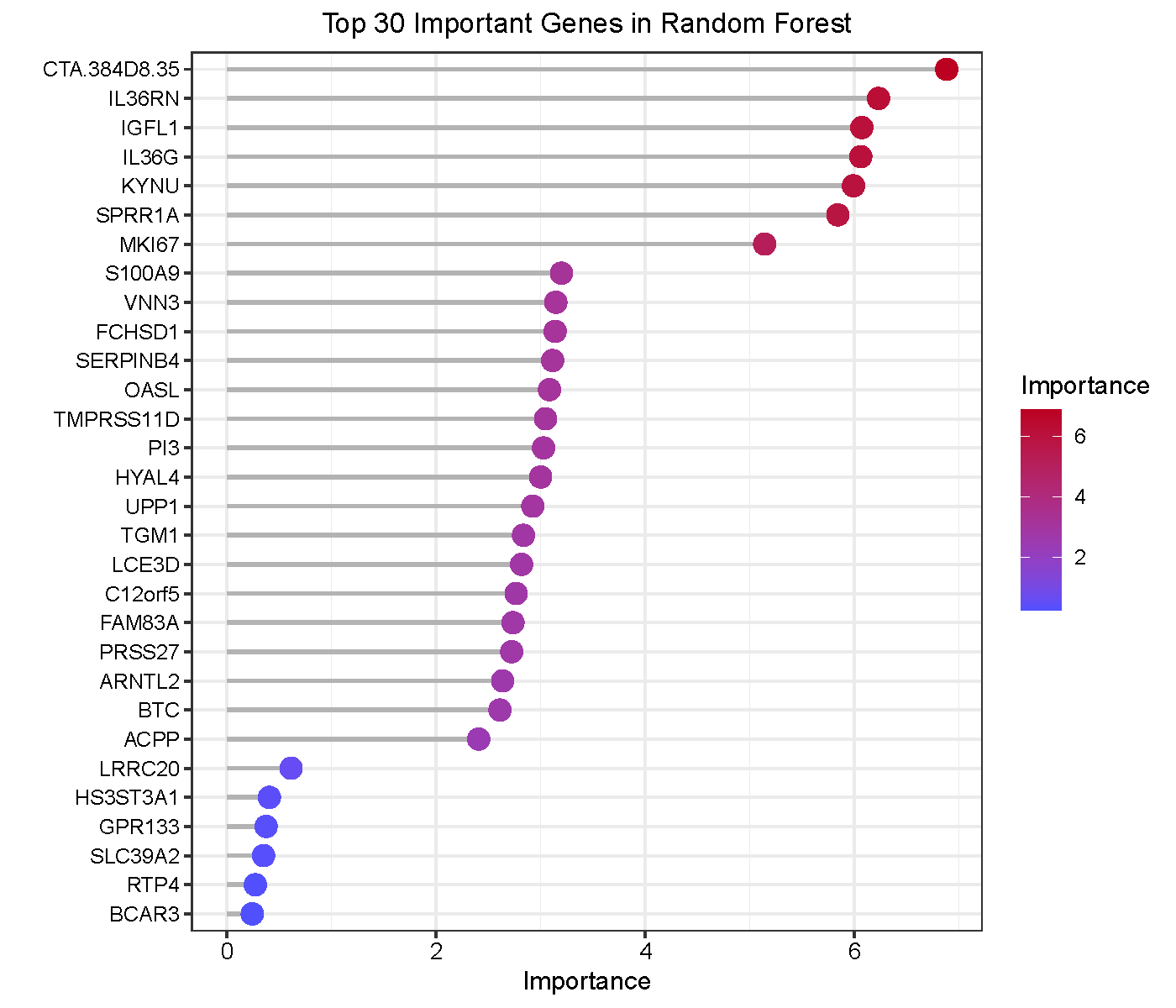
## 15. 绘制Top30基因重要性图
##############################
plot_df <- importance_df
if (nrow(plot_df) > 30) {
plot_df <- plot_df[1:30, , drop = FALSE]
}
plot_df$Gene <- factor(plot_df$Gene, levels = rev(plot_df$Gene))
plot_df$Importance <- as.numeric(plot_df$Importance)
p <- ggplot(plot_df, aes(x = Importance, y = Gene)) +
geom_segment(
aes(x = 0, xend = Importance, y = Gene, yend = Gene),
color = "grey70",
linewidth = 0.8
) +
geom_point(aes(color = Importance), size = 4) +
scale_colour_gradient(low = "#5050FFFF", high = "#BB0021FF") +
labs(
title = "Top 30 Important Genes in Random Forest",
x = "Importance",
y = ""
) +
theme_bw() +
theme(
plot.title = element_text(hjust = 0.5, size = 12),
axis.text.x = element_text(color = "black", size = 10),
axis.text.y = element_text(color = "black", size = 9)
)
pdf(file = "geneImportance_Top1000.pdf", width = 7, height = 6)
tryCatch({
print(p)
}, error = function(e) {
plot.new()
text(0.5, 0.5, paste("Plot failed:", e$message))
})
dev.off()
##############################
## 16. 控制台输出总结
##############################
cat("\n=============================\n")
cat("RF 分析完成\n")
cat("=============================\n")
cat("最优树数:", optionTrees, "\n")
cat("最小OOB误差:", min(oob_error, na.rm = TRUE), "\n")
cat("最终基因数:", length(final_genes), "\n")
cat("最终结果中目标基因:", paste(intersect(target_genes, final_genes), collapse = ", "), "\n")
cat("输出文件:\n")
cat("1. forest_Top1000.pdf\n")
cat("2. geneImportance_Top1000.pdf\n")
cat("3. RF.final.genes.txt\n")
cat("=============================\n")
#======================================
# 原创代码无删减,编写不易,论文使用本代码绘图,请引用:www.tcmbiohub.com
# 祝大家投稿顺利!
 TCM Bio Hub⠀⠀⠀
TCM Bio Hub⠀⠀⠀