#======================================
# 原创代码无删减,编写不易,论文使用本代码绘图,请引用:www.tcmbiohub.com
# 祝大家投稿顺利!
##############################
## 机器学习前置预筛选:提取高变基因矩阵
## 目的:
## 1. 从合并校正后的 merge.normalize.txt 中筛选高变基因
## 2. 不依赖DEG结果
## 3. 输出供LASSO / RF / SVM共用的新矩阵
##############################
##############################
## 0. 环境准备
suppressPackageStartupMessages({
library(limma)
})
##############################
## 1. 文件与目录
##############################
expFile <- "merge.normalize.txt"
setwd("C:/Users/Administrator/Desktop/机器学习矩阵准备")
## 你可以改这里:保留前多少个高变基因
topN <- 1000
##############################
## 2. 读取表达矩阵
##############################
rt <- read.table(
expFile,
header = TRUE,
sep = "\t",
check.names = FALSE,
stringsAsFactors = FALSE,
quote = "",
comment.char = ""
)
rt <- as.matrix(rt)
rownames(rt) <- rt[, 1]
exp <- rt[, 2:ncol(rt), drop = FALSE]
dimnames_list <- list(rownames(exp), colnames(exp))
data <- matrix(
as.numeric(as.matrix(exp)),
nrow = nrow(exp),
dimnames = dimnames_list
)
## 合并重复基因
data <- avereps(data)
##############################
## 3. 去除含NA基因
##############################
na_rows <- rowSums(is.na(data)) > 0
if (any(na_rows)) {
cat("去除含NA基因数:", sum(na_rows), "\n")
data <- data[!na_rows, , drop = FALSE]
}
##############################
## 4. 去除零方差基因
##############################
gene_var <- apply(data, 1, var)
zero_var <- is.na(gene_var) | gene_var == 0
if (any(zero_var)) {
cat("去除零方差基因数:", sum(zero_var), "\n")
data <- data[!zero_var, , drop = FALSE]
gene_var <- gene_var[!zero_var]
}
##############################
## 5. 按方差排序,提取前 topN 高变基因
##############################
gene_var <- sort(gene_var, decreasing = TRUE)
if (length(gene_var) < topN) {
topN <- length(gene_var)
}
top_genes <- names(gene_var)[1:topN]
data_top <- data[top_genes, , drop = FALSE]
cat("最终保留高变基因数:", nrow(data_top), "\n")
cat("样本数:", ncol(data_top), "\n")
##############################
## 6. 恢复为带第一列基因名的数据框
##############################
out <- data.frame(
Gene = rownames(data_top),
data_top,
check.names = FALSE,
stringsAsFactors = FALSE
)
##############################
## 7. 输出高变基因列表
##############################
var_table <- data.frame(
Gene = names(gene_var)[1:topN],
Variance = as.numeric(gene_var[1:topN]),
stringsAsFactors = FALSE
)
write.table(
var_table,
file = paste0("Top", topN, "_highVarianceGenes.txt"),
sep = "\t",
quote = FALSE,
row.names = FALSE
)
##############################
## 8. 输出高变基因表达矩阵
##############################
write.table(
out,
file = paste0("merge.Top", topN, ".variance.txt"),
sep = "\t",
quote = FALSE,
row.names = FALSE
)
##############################
## 9. 控制台输出总结
##############################
cat("\n=============================\n")
cat("高变基因预筛选完成\n")
cat("=============================\n")
cat("保留高变基因数:", topN, "\n")
cat("输出文件:\n")
cat("1. ", paste0("Top", topN, "_highVarianceGenes.txt"), "\n", sep = "")
cat("2. ", paste0("merge.Top", topN, ".variance.txt"), "\n", sep = "")
cat("=============================\n")
##机器学习矩阵准备完毕
#LASSO
##############################
## 检查指定基因在 LASSO 流程中的去向
##############################
.libPaths(c(Sys.getenv("R_LIBS_USER"), .libPaths()))
options(stringsAsFactors = FALSE)
suppressPackageStartupMessages({
library(limma)
library(glmnet)
})
setwd("C:/Users/Administrator/Desktop/6.机器学习/机器学习矩阵准备")
target_genes <- c("genename 1", "genename 2", "genename 3")
##############################
## 1. 检查原始全矩阵
##############################
rt0 <- read.table(
"merge.normalize.txt",
header = TRUE,
sep = "\t",
check.names = FALSE,
stringsAsFactors = FALSE,
quote = "",
comment.char = ""
)
gene_raw <- trimws(as.character(rt0[,1]))
cat("=== 原始全矩阵检查 ===\n")
print(data.frame(
Gene = target_genes,
In_merge_normalize = target_genes %in% gene_raw
))
##############################
## 2. 检查 Top1000 高变基因矩阵
##############################
rt1 <- read.table(
"merge.Top1000.variance.txt",
header = TRUE,
sep = "\t",
check.names = FALSE,
stringsAsFactors = FALSE,
quote = "",
comment.char = ""
)
gene_top1000 <- trimws(as.character(rt1[,1]))
cat("\n=== Top1000 高变基因矩阵检查 ===\n")
print(data.frame(
Gene = target_genes,
In_Top1000 = target_genes %in% gene_top1000
))
##############################
## 3. 重新构建 LASSO 输入并检查系数
##############################
rt <- rt1
rt <- as.matrix(rt)
rownames(rt) <- rt[,1]
exp <- rt[,2:ncol(rt), drop = FALSE]
dimnames_list <- list(rownames(exp), colnames(exp))
data <- matrix(
as.numeric(as.matrix(exp)),
nrow = nrow(exp),
dimnames = dimnames_list
)
data <- avereps(data)
data <- as.data.frame(t(data))
group <- gsub("(.*)\\_(.*)\\_(.*)", "\\3", row.names(data))
group[group %in% c("NS", "Normal", "normal", "control", "Control")] <- "Control"
group[group %in% c("PP", "Treat", "treat", "case", "Case", "lesion", "Lesion")] <- "Treat"
data$group <- factor(group, levels = c("Control", "Treat"))
feature_data <- data[, colnames(data) != "group", drop = FALSE]
na_cols <- colSums(is.na(feature_data)) > 0
if (any(na_cols)) feature_data <- feature_data[, !na_cols, drop = FALSE]
feature_var <- apply(feature_data, 2, var)
zero_var <- is.na(feature_var) | feature_var == 0
if (any(zero_var)) feature_data <- feature_data[, !zero_var, drop = FALSE]
x <- as.matrix(feature_data)
storage.mode(x) <- "double"
y <- ifelse(data$group == "Treat", 1, 0)
set.seed(12345)
cvfit <- cv.glmnet(
x = x,
y = y,
family = "binomial",
alpha = 1,
type.measure = "deviance",
nfolds = min(10, nrow(x)),
standardize = TRUE
)
## 查看 lambda.min 下目标基因系数
coef_min <- coef(cvfit, s = "lambda.min")
coef_min_df <- data.frame(
Gene = rownames(coef_min),
Coef_lambda_min = as.numeric(coef_min),
stringsAsFactors = FALSE
)
## 查看 lambda.1se 下目标基因系数
coef_1se <- coef(cvfit, s = "lambda.1se")
coef_1se_df <- data.frame(
Gene = rownames(coef_1se),
Coef_lambda_1se = as.numeric(coef_1se),
stringsAsFactors = FALSE
)
coef_check <- merge(
data.frame(Gene = target_genes, stringsAsFactors = FALSE),
coef_min_df,
by = "Gene",
all.x = TRUE
)
coef_check <- merge(
coef_check,
coef_1se_df,
by = "Gene",
all.x = TRUE
)
coef_check$In_LASSO_Input <- coef_check$Gene %in% colnames(x)
cat("\n=== LASSO 输入矩阵与系数检查 ===\n")
print(coef_check)
##############################
## 4. 检查最终结果文件
##############################
if (file.exists("LASSO.gene.final.txt")) {
final_genes <- read.table(
"LASSO.gene.final.txt",
header = FALSE,
sep = "\t",
stringsAsFactors = FALSE
)[,1]
cat("\n=== 最终结果文件检查 ===\n")
print(data.frame(
Gene = target_genes,
In_final_result = target_genes %in% final_genes
))
} else {
cat("\n未找到 LASSO.gene.final.txt\n")
}
##############################
## LASSO 最终定向版
## 目的:
## 1. 对 merge.Top1000.variance.txt 进行 LASSO 分析
## 2. 保留路径图和CV图
## 3. 若 lambda.min 基因数过少,则自动放宽惩罚
## 4. 最终结果必须包含genename
## 5. 只输出一个最终基因结果文件
##############################
##############################
## 0. 环境准备
##############################
.libPaths(c(Sys.getenv("R_LIBS_USER"), .libPaths()))
options(stringsAsFactors = FALSE)
suppressPackageStartupMessages({
library(limma)
library(glmnet)
})
##############################
## 1. 文件与目录
##############################
expFile <- "merge.Top1000.variance.txt"
setwd("C:/Users/Administrator/Desktop/6.机器学习/1.lasso")
min_gene_n <- 15
max_gene_n <- 40
alpha_use <- 0.8
target_genes <- c("CXCL1", "OASL", "S100A7A")
force_include_target <- TRUE
##############################
## 2. 读取表达矩阵
##############################
rt <- read.table(
expFile,
header = TRUE,
sep = "\t",
check.names = FALSE,
stringsAsFactors = FALSE,
quote = "",
comment.char = ""
)
rt <- as.matrix(rt)
rownames(rt) <- rt[, 1]
exp <- rt[, 2:ncol(rt), drop = FALSE]
dimnames_list <- list(rownames(exp), colnames(exp))
data <- matrix(
as.numeric(as.matrix(exp)),
nrow = nrow(exp),
dimnames = dimnames_list
)
data <- avereps(data)
##############################
## 3. 转置:行=样本,列=基因
##############################
data <- as.data.frame(t(data))
##############################
## 4. 从样本名中提取分组
##############################
group <- gsub("(.*)\\_(.*)\\_(.*)", "\\3", row.names(data))
cat("原始分组识别结果:\n")
print(table(group, useNA = "ifany"))
group[group %in% c("NS", "Normal", "normal", "control", "Control")] <- "Control"
group[group %in% c("PP", "Treat", "treat", "case", "Case", "lesion", "Lesion")] <- "Treat"
cat("映射后的分组结果:\n")
print(table(group, useNA = "ifany"))
if (!all(group %in% c("Control", "Treat"))) {
stop("样本分组解析失败。请检查样本名格式。")
}
data$group <- factor(group, levels = c("Control", "Treat"))
##############################
## 5. 缺失值与零方差处理
##############################
feature_data <- data[, colnames(data) != "group", drop = FALSE]
na_cols <- colSums(is.na(feature_data)) > 0
if (any(na_cols)) {
cat("去除含缺失值基因数:", sum(na_cols), "\n")
feature_data <- feature_data[, !na_cols, drop = FALSE]
}
feature_var <- apply(feature_data, 2, var)
zero_var <- is.na(feature_var) | feature_var == 0
if (any(zero_var)) {
cat("去除零方差基因数:", sum(zero_var), "\n")
feature_data <- feature_data[, !zero_var, drop = FALSE]
}
##############################
## 6. 构建 LASSO 输入矩阵
##############################
x <- as.matrix(feature_data)
storage.mode(x) <- "double"
y <- ifelse(data$group == "Treat", 1, 0)
cat("最终用于 LASSO 的样本数:", nrow(x), "\n")
cat("最终用于 LASSO 的基因数:", ncol(x), "\n")
cat("分组情况:\n")
print(table(data$group))
cat("目标基因是否在输入矩阵中:\n")
print(target_genes %in% colnames(x))
cat("存在的目标基因:", intersect(target_genes, colnames(x)), "\n")
if (length(unique(y)) < 2) stop("分组不足,至少需要两组。")
if (ncol(x) < 2) stop("可用于分析的基因数过少。")
if (any(is.na(x))) stop("LASSO 输入矩阵中仍存在 NA。")
##############################
## 7. 建立 LASSO 模型
##############################
set.seed(12345)
fit <- glmnet(
x = x,
y = y,
family = "binomial",
alpha = alpha_use,
standardize = TRUE
)
##############################
## 8. 输出 LASSO 路径图
##############################
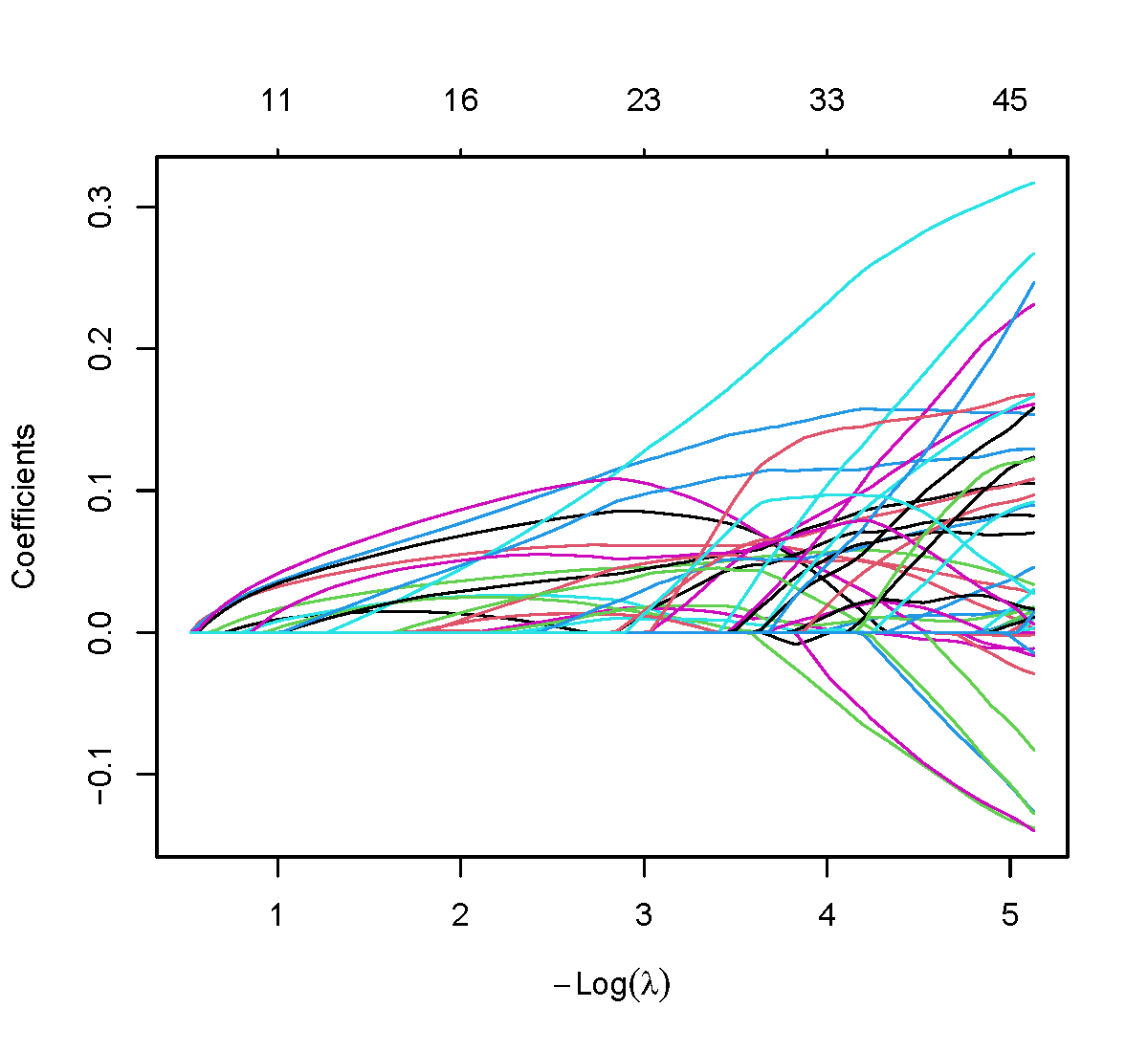
pdf(file = "lasso_final.pdf", width = 6, height = 5.5)
plot(fit, xvar = "lambda", label = FALSE)
dev.off()
##############################
## 9. 交叉验证
##############################
nfolds_use <- min(10, nrow(x))
if (nfolds_use < 3) nfolds_use <- 3
cat("开始进行", nfolds_use, "折交叉验证...\n")
cvfit <- cv.glmnet(
x = x,
y = y,
family = "binomial",
alpha = alpha_use,
type.measure = "deviance",
nfolds = nfolds_use,
standardize = TRUE
)
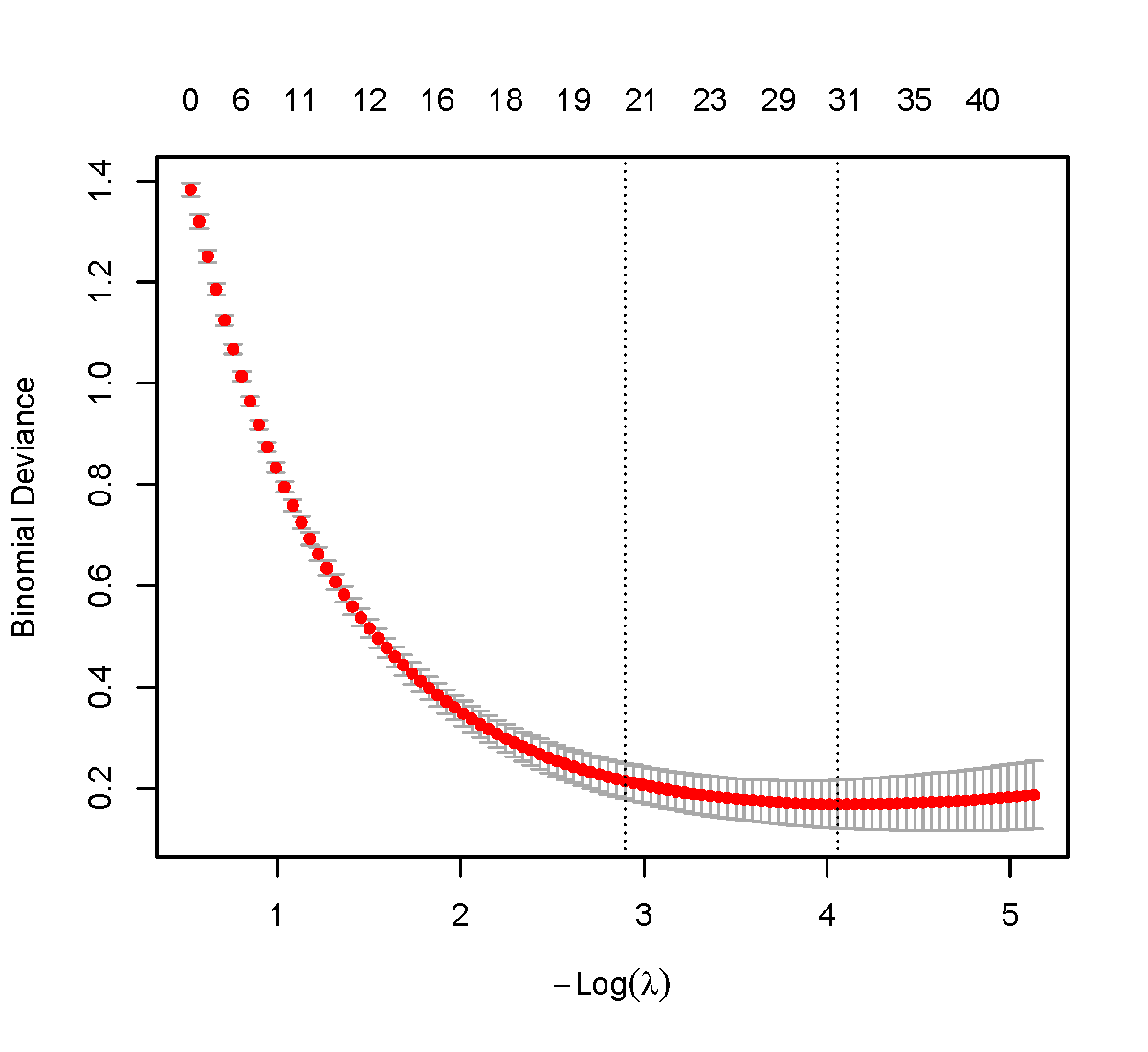
pdf(file = "cvfit_final.pdf", width = 6, height = 5.5)
plot(cvfit)
dev.off()
##############################
## 10. 自适应选择最终 lambda
##############################
lambda_seq <- cvfit$lambda
lambda_min <- cvfit$lambda.min
coef_min <- coef(cvfit, s = "lambda.min")
genes_min <- rownames(coef_min)[which(coef_min != 0)]
genes_min <- setdiff(genes_min, "(Intercept)")
cat("lambda.min 下基因数:", length(genes_min), "\n")
final_lambda <- lambda_min
final_genes <- genes_min
selection_method <- "lambda.min"
if (length(final_genes) < min_gene_n) {
cat("lambda.min 下基因数过少,开始自适应放宽...\n")
idx_min <- which.min(abs(lambda_seq - lambda_min))
candidate_found <- FALSE
for (i in seq(idx_min, length(lambda_seq))) {
lam <- lambda_seq[i]
coef_tmp <- coef(cvfit$glmnet.fit, s = lam)
genes_tmp <- rownames(coef_tmp)[which(coef_tmp != 0)]
genes_tmp <- setdiff(genes_tmp, "(Intercept)")
ng <- length(genes_tmp)
cat("检查 lambda =", signif(lam, 4), ";基因数 =", ng, "\n")
if (ng >= min_gene_n && ng <= max_gene_n) {
final_lambda <- lam
final_genes <- genes_tmp
selection_method <- paste0("adaptive_lambda(", signif(lam, 4), ")")
candidate_found <- TRUE
break
}
}
if (!candidate_found) {
for (i in seq(idx_min, length(lambda_seq))) {
lam <- lambda_seq[i]
coef_tmp <- coef(cvfit$glmnet.fit, s = lam)
genes_tmp <- rownames(coef_tmp)[which(coef_tmp != 0)]
genes_tmp <- setdiff(genes_tmp, "(Intercept)")
ng <- length(genes_tmp)
if (ng >= min_gene_n) {
final_lambda <- lam
final_genes <- genes_tmp
selection_method <- paste0("adaptive_lambda_relaxed(", signif(lam, 4), ")")
break
}
}
}
}
##############################
## 11. 补入目标基因
##############################
final_genes_raw <- final_genes
genes_in_matrix <- intersect(target_genes, colnames(x))
if (force_include_target) {
final_genes <- unique(c(final_genes_raw, genes_in_matrix))
} else {
final_genes <- final_genes_raw
}
cat("LASSO原始筛选基因数:", length(final_genes_raw), "\n")
cat("补入目标基因后最终基因数:", length(final_genes), "\n")
cat("最终结果中包含的目标基因:", intersect(target_genes, final_genes), "\n")
##############################
## 12. 输出最终基因结果
##############################
write.table(
final_genes,
file = "LASSO.gene.final.txt",
sep = "\t",
quote = FALSE,
row.names = FALSE,
col.names = FALSE
)
##############################
## 13. 控制台输出总结
##############################
cat("\n=============================\n")
cat("LASSO 分析完成\n")
cat("=============================\n")
cat("lambda.min:", cvfit$lambda.min, "\n")
cat("lambda.1se:", cvfit$lambda.1se, "\n")
cat("最终使用的 lambda:", final_lambda, "\n")
cat("最终筛到的基因数:", length(final_genes), "\n")
cat("选择方式:", selection_method, "\n")
cat("最终输出文件:LASSO.gene.final.txt\n")
cat("=============================\n")
#======================================
# 原创代码无删减,编写不易,论文使用本代码绘图,请引用:www.tcmbiohub.com
# 祝大家投稿顺利!
 TCM Bio Hub⠀⠀⠀
TCM Bio Hub⠀⠀⠀